JunctionViewer: customizable annotation software for repeat-rich genomic regions
Description
JunctionViewer generates concise graphical representations of DNA sequence features using NCBI BLAST/WU-BLAST, cross_match, and MUMmer, as well as customizable annotation data. Results are displayed through the graphical interface or can be viewed from PostScript® output. It is particularly suited for annotating repeat-rich genomic regions, such as centromeres. The name "JunctionViewer" stems from the software's original purpose - visualizing "repeat junctions" in the centromeres of maize (Luce et al., 2006).
For more details and if you use this software please refer to the related publication:
Wolfgruber T, Presting G (2010) JunctionViewer: customizable annotation software for repeat-rich genomic regions. BMC Bioinformatics 11: 23. doi:10.1186/1471-2105-11-23. url:http://www.biomedcentral.com/1471-2105/11/23
Features
- User definable parameters for each BLAST/cross_match database.
- Competes all BLAST HSPs/cross_match results between subject databases to reveal only the best results.
- Displays exact sequence matches within query sequence as reported by MUMmer.
- Plots customizable annotation data (e.g., ChIP-Seq mapped reads positions) on top of BLAST/cross_match/MUMmer results.
Usage
- Run JunctionViewer script without parameters to generate a template parameters file.
- Populate parameters file (comments are given indicating how this should be done).
- Run JunctionViewer with parameters file as argument.
Requirements
Perl, Perl modules BioPerl and Tk, NCBI BLAST and/or WU-BLAST, cross_match, and MUMmer.
Downloading
Download the JunctionViewer package through SourceForge.net®. This package includes the software, setup/usage instructions, example files, and license documents:
JunctionViewer-*.pl = Software file
README-*.txt = Setup, usage, testing, and known issues are described in these files for linux/Windows users
JunctionViewer-*example_parameters-[linux,windows].txt = Example parameters files for linux/windows users
example_* = Example files for use with example parameters files
COPYING-[linux,windows].txt = GPLv3 software license documents for linux/windows users
Running JunctionViewer using example files
- Install required Perl software/Perl modules by reading the README-linux.txt file if you are using a unix-like OS, or README-windows.txt if you are a Windows user.
- Obtain at least NCBI BLAST. Linux users may also obtain cross_match and/or MUMmer. If you are unable to get a binary, e.g., cross_match, annotations using that algorithm may be omitted (see step 3).
- Re-define BLAST/cross_match/MUMmer binary locations in example parameters file where indicated under comment lines "# !!!Change the following to use your own binary location!!!" If you were unable to get a binary, you may also set a binary to no value ("Binary="), which will mean alignments using that binary will be skipped.
- Run the script with the updated parameters file, i.e., using command line statement 'JunctionViewer-xx.pl JunctionViewer-xx.pl.example_parameters-[linux,windows].txt'.
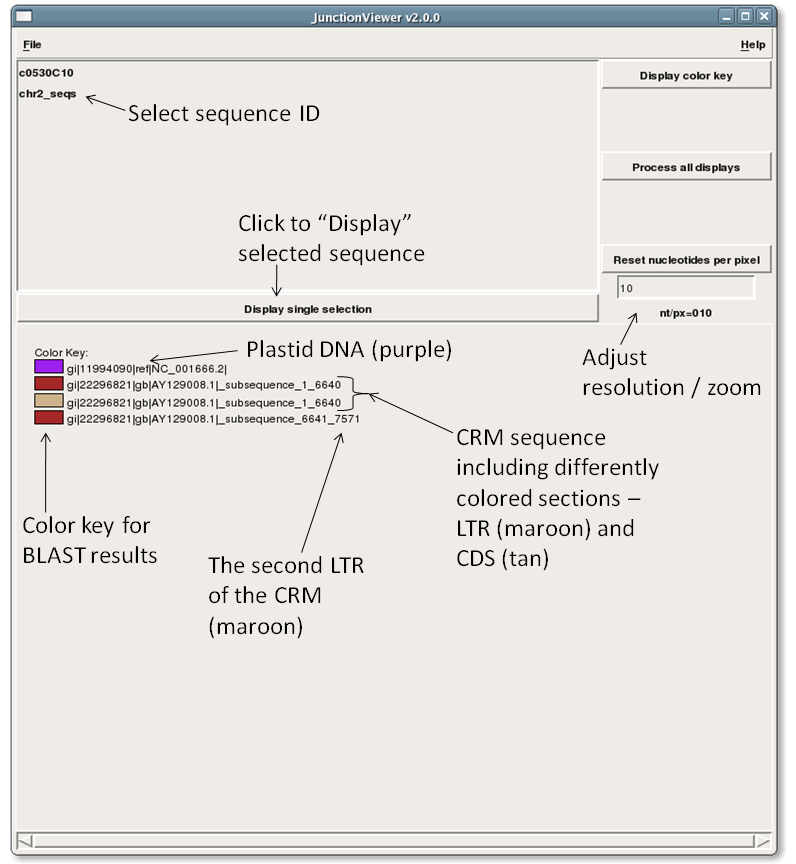
- Select a sequence and press "Display single selection" to visualize its features.
Results from example files
| GUI |
Query sequence #1 ("c0530C10") |
Query sequence #2 ("chr2_seqs") |
 |
 |
 |
License
GPLv3
Copyright 2010,2009 Thomas K. Wolfgruber